Publications
2026
- JGR
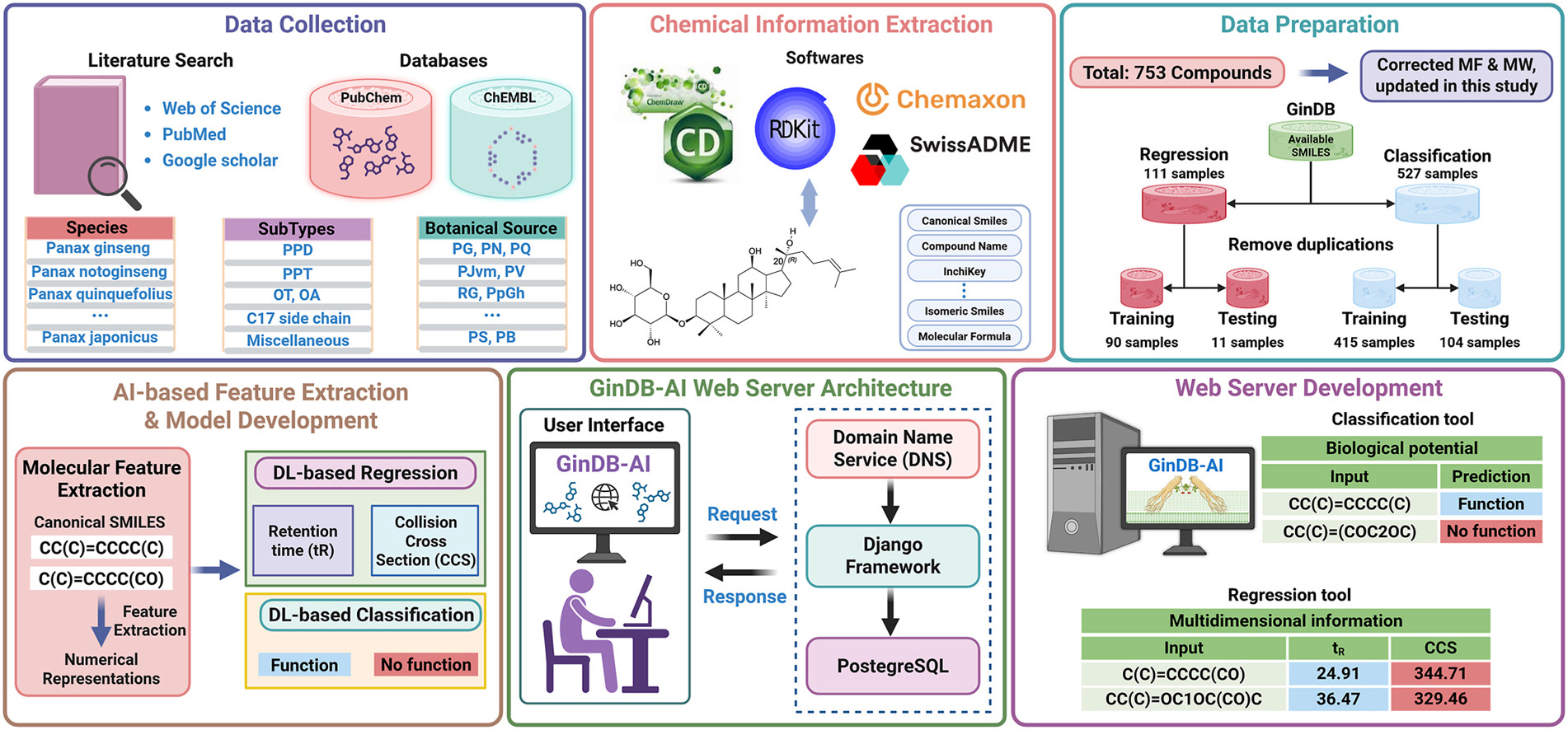 GinDB-AI: An integrated ginsenoside database and AI-driven platform for multidimensional information and biological activity predictionNguyen Doan Hieu Nguyen, Vinoth Kumar Sangaraju, Duong Thanh Tran, Nhat Truong Pham, Jae Youl Cho, and Balachandran ManavalanJournal of Ginseng Research, 2026
GinDB-AI: An integrated ginsenoside database and AI-driven platform for multidimensional information and biological activity predictionNguyen Doan Hieu Nguyen, Vinoth Kumar Sangaraju, Duong Thanh Tran, Nhat Truong Pham, Jae Youl Cho, and Balachandran ManavalanJournal of Ginseng Research, 2026@article{nguyen2026gindb, title = {GinDB-AI: An integrated ginsenoside database and AI-driven platform for multidimensional information and biological activity prediction}, author = {Nguyen, Nguyen Doan Hieu and Sangaraju, Vinoth Kumar and Tran, Duong Thanh and Pham, Nhat Truong and Cho, Jae Youl and Manavalan, Balachandran}, journal = {Journal of Ginseng Research}, pages = {100986}, year = {2026}, issn = {1226-8453}, doi = {doi.org/10.1016/j.jgr.2026.100986}, url = {https://www.sciencedirect.com/science/article/pii/S1226845326000126}, published = {true}, dimensions = {true}, }
2024
- CBM
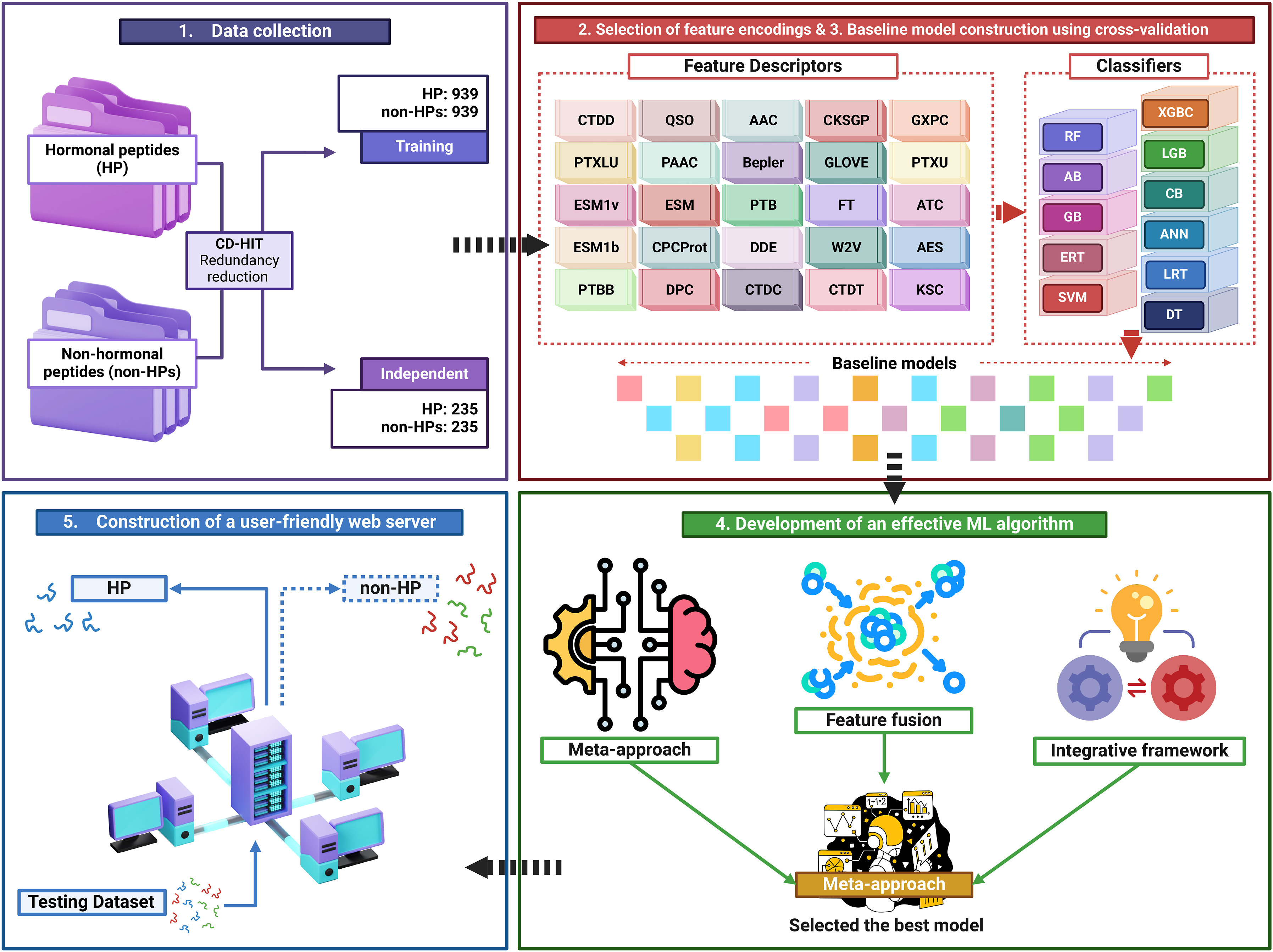 mHPpred: Accurate identification of peptide hormones using multi-view feature learningShaherin Basith, Vinoth Kumar Sangaraju, Balachandran Manavalan, and Gwang LeeComputers in Biology and Medicine, 2024
mHPpred: Accurate identification of peptide hormones using multi-view feature learningShaherin Basith, Vinoth Kumar Sangaraju, Balachandran Manavalan, and Gwang LeeComputers in Biology and Medicine, 2024Peptide hormones were first used in medicine in the early 20th century, with the pivotal event being the isolation and purification of insulin in 1921. These hormones are integral to a sophisticated system that emerged early in evolution to regulate growth, development, and homeostasis. They serve as targeted signaling molecules that transfer specific information between cells and organs, ensuring coordinated and precise physiological responses. While experimental methods for identifying peptide hormones present challenges such as low abundance, stability issues, and complexity, computational methods offer promising alternatives. Advances in machine learning and bioinformatics have facilitated the prediction of peptide hormones, further enhancing their therapeutic potential. In this study, we explored three different computational frameworks for peptide hormone identification and determined that the meta-approach was the most suitable. Firstly, we evaluated the discriminative power of 26 feature descriptors using a series of baseline models and identified seven feature descriptors with high predictive potential. Through a systematic approach, we then selected the top 20 performing baseline models and integrated their predicted probabilities to train a meta-model, leveraging the strengths of multiple prediction strategies. Our final light gradient boosting-based meta-model, mHPpred, significantly outperformed the existing method, HOPPred, on both benchmarking and independent datasets. Notably, mHPpred also demonstrated superior performance compared to the hybrid and integrative framework approaches employed in this study. This superiority demonstrates the effectiveness of our multi-view feature learning strategy in capturing discriminative features and providing a more accurate prediction model for peptide hormones. mHPpred is publicly accessible at: https://balalab-skku.org/mHPpred.
@article{BASITH2024109297, title = {mHPpred: Accurate identification of peptide hormones using multi-view feature learning}, author = {Basith, Shaherin and Sangaraju, Vinoth Kumar and Manavalan, Balachandran and Lee, Gwang}, journal = {Computers in Biology and Medicine}, volume = {183}, pages = {109297}, year = {2024}, issn = {0010-4825}, doi = {10.1016/j.compbiomed.2024.109297}, url = {https://www.sciencedirect.com/science/article/pii/S0010482524013829}, published = {true}, dimensions = {true}, } - JMB
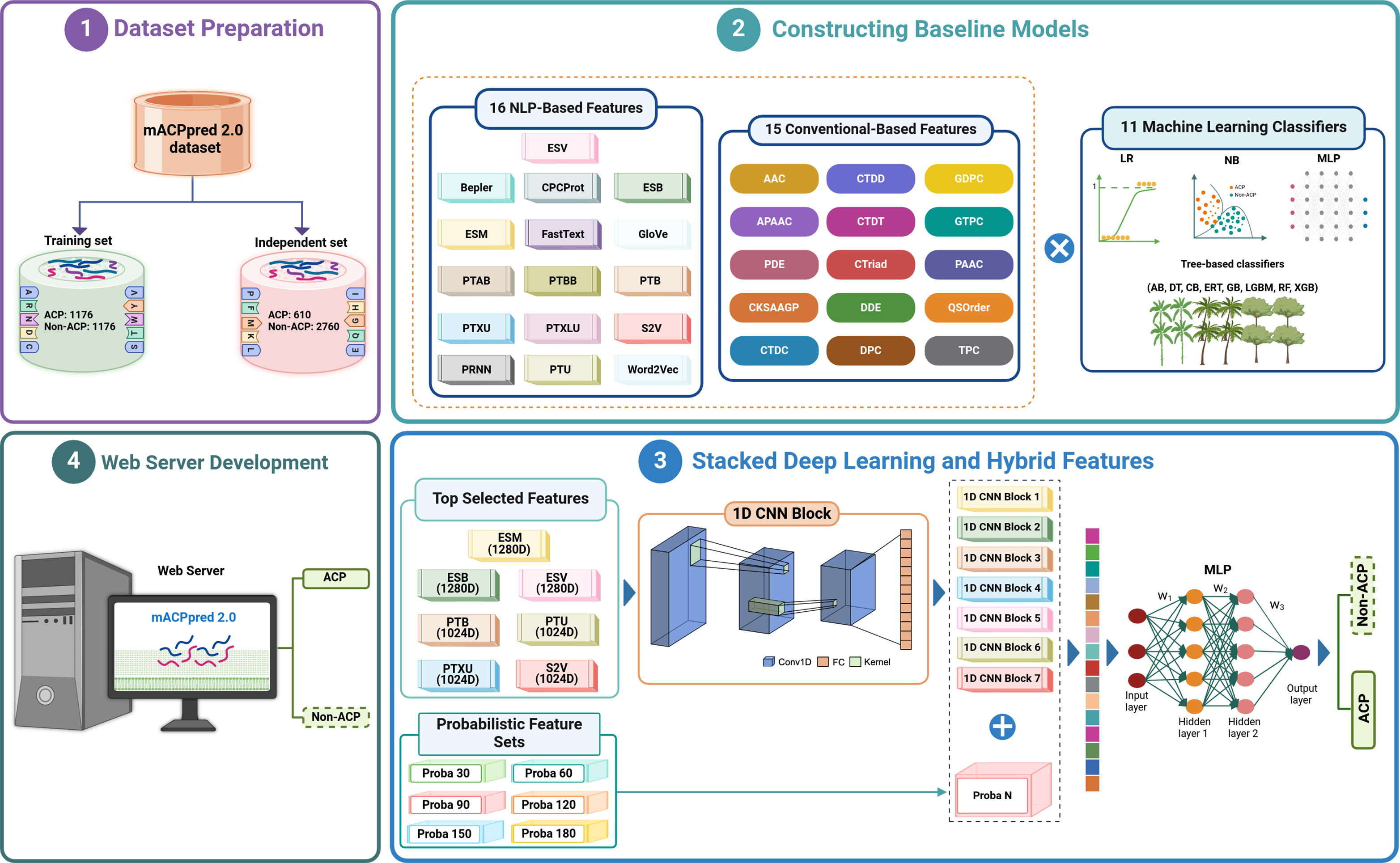 mACPpred 2.0: Stacked deep learning for anticancer peptide prediction with integrated spatial and probabilistic feature representationsVinoth Kumar Sangaraju, Nhat Truong Pham, Leyi Wei, Xue Yu, and Balachandran ManavalanJournal of Molecular Biology, 2024
mACPpred 2.0: Stacked deep learning for anticancer peptide prediction with integrated spatial and probabilistic feature representationsVinoth Kumar Sangaraju, Nhat Truong Pham, Leyi Wei, Xue Yu, and Balachandran ManavalanJournal of Molecular Biology, 2024Anticancer peptides (ACPs), naturally occurring molecules with remarkable potential to target and kill cancer cells. However, identifying ACPs based solely from their primary amino acid sequences remains a major hurdle in immunoinformatics. In the past, several web-based machine learning (ML) tools have been proposed to assist researchers in identifying potential ACPs for further testing. Notably, our meta-approach method, mACPpred, introduced in 2019, has significantly advanced the field of ACP research. Given the exponential growth in the number of characterized ACPs, there is now a pressing need to create an updated version of mACPpred. To develop mACPpred 2.0, we constructed an up-to-date benchmarking dataset by integrating all publicly available ACP datasets. We employed a large-scale of feature descriptors, encompassing both conventional feature descriptors and advanced pre-trained natural language processing (NLP)-based embeddings. We evaluated their ability to discriminate between ACPs and non-ACPs using eleven different classifiers. Subsequently, we employed a stacked deep learning (SDL) approach, incorporating 1D convolutional neural network (1D CNN) blocks and hybrid features. These features included the top seven performing NLP-based features and 90 probabilistic features, allowing us to identify hidden patterns within these diverse features and improve the accuracy of our ACP prediction model. This is the first study to integrate spatial and probabilistic feature representations for predicting ACPs. Rigorous cross-validation and independent tests conclusively demonstrated that mACPpred 2.0 not only surpassed its predecessor (mACPpred) but also outperformed the existing state-of-the-art predictors, highlighting the importance of advanced feature representation capabilities attained through SDL. To facilitate widespread use and accessibility, we have developed a user-friendly for mACPpred 2.0, available at https://balalab-skku.org/mACPpred2/.
@article{sangaraju2024macppred, title = {mACPpred 2.0: Stacked deep learning for anticancer peptide prediction with integrated spatial and probabilistic feature representations}, author = {Sangaraju, Vinoth Kumar and Pham, Nhat Truong and Wei, Leyi and Yu, Xue and Manavalan, Balachandran}, journal = {Journal of Molecular Biology}, volume = {436}, number = {17}, pages = {168687}, year = {2024}, publisher = {Elsevier}, doi = {10.1016/j.jmb.2024.168687}, url = {https://www.sciencedirect.com/science/article/pii/S0022283624002894}, published = {true}, dimensions = {true}, }
2023
- CBM
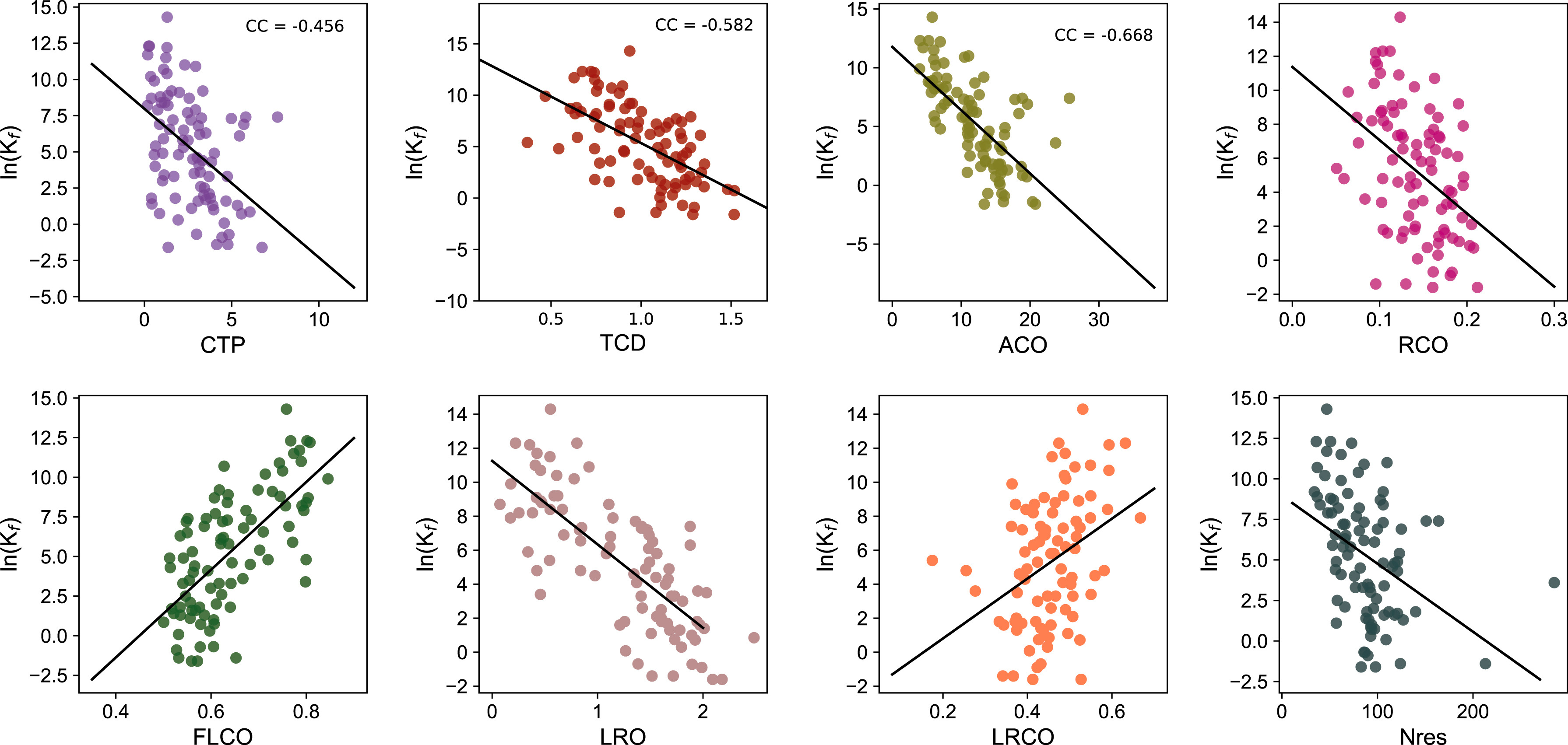 Computational prediction of protein folding rate using structural parameters and network centrality measuresSaraswathy Nithiyanandam, Vinoth Kumar Sangaraju, Balachandran Manavalan, and Gwang LeeComputers in Biology and Medicine, 2023
Computational prediction of protein folding rate using structural parameters and network centrality measuresSaraswathy Nithiyanandam, Vinoth Kumar Sangaraju, Balachandran Manavalan, and Gwang LeeComputers in Biology and Medicine, 2023Protein folding is a complex physicochemical process whereby a polymer of amino acids samples numerous conformations in its unfolded state before settling on an essentially unique native three-dimensional (3D) structure. To understand this process, several theoretical studies have used a set of 3D structures, identified different structural parameters, and analyzed their relationships using the natural logarithmic protein folding rate (ln(kf)). Unfortunately, these structural parameters are specific to a small set of proteins that are not capable of accurately predicting ln(kf) for both two-state (TS) and non-two-state (NTS) proteins. To overcome the limitations of the statistical approach, a few machine learning (ML)-based models have been proposed using limited training data. However, none of these methods can explain plausible folding mechanisms. In this study, we evaluated the predictive capabilities of ten different ML algorithms using eight different structural parameters and five different network centrality measures based on newly constructed datasets. In comparison to the other nine regressors, support vector machine was found to be the most appropriate for predicting ln(kf) with mean absolute differences of 1.856, 1.55, and 1.745 for the TS, NTS, and combined datasets, respectively. Furthermore, combining structural parameters and network centrality measures improves the prediction performance compared to individual parameters, indicating that multiple factors are involved in the folding process.
@article{nithiyanandam2023computational, title = {Computational prediction of protein folding rate using structural parameters and network centrality measures}, author = {Nithiyanandam, Saraswathy and Sangaraju, Vinoth Kumar and Manavalan, Balachandran and Lee, Gwang}, journal = {Computers in Biology and Medicine}, volume = {155}, pages = {106436}, year = {2023}, publisher = {Elsevier}, doi = {10.1016/j.compbiomed.2022.106436}, url = {https://www.sciencedirect.com/science/article/pii/S0010482522011441}, published = {true}, dimensions = {true}, } - Research
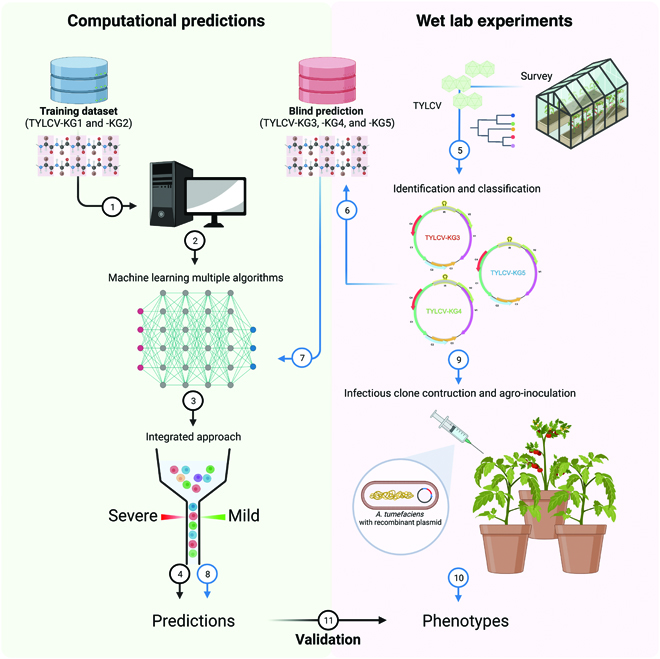 An effective integrated machine learning framework for identifying severity of tomato yellow leaf curl virus and their experimental validationNattanong Bupi, Vinoth Kumar Sangaraju, Le Thi Phan, Aamir Lal, Thuy Thi Bich Vo, Phuong Thi Ho, Muhammad Amir Qureshi, Marjia Tabassum, Sukchan Lee, and Balachandran ManavalanResearch, 2023
An effective integrated machine learning framework for identifying severity of tomato yellow leaf curl virus and their experimental validationNattanong Bupi, Vinoth Kumar Sangaraju, Le Thi Phan, Aamir Lal, Thuy Thi Bich Vo, Phuong Thi Ho, Muhammad Amir Qureshi, Marjia Tabassum, Sukchan Lee, and Balachandran ManavalanResearch, 2023Tomato yellow leaf curl virus (TYLCV) dispersed across different countries, specifically to subtropical regions, associated with more severe symptoms. Since TYLCV was first isolated in 1931, it has been a menace to tomato industrial production worldwide over the past century. Three groups were newly isolated from TYLCV-resistant tomatoes in 2022; however, their functions are unknown. The development of machine learning (ML)-based models using characterized sequences and evaluating blind predictions is one of the major challenges in interdisciplinary research. The purpose of this study was to develop an integrated computational framework for the accurate identification of symptoms (mild or severe) based on TYLCV sequences (isolated in Korea). For the development of the framework, we first extracted 11 different feature encodings and hybrid features from the training data and then explored 8 different classifiers and developed their respective prediction models by using randomized 10-fold cross-validation. Subsequently, we carried out a systematic evaluation of these 96 developed models and selected the top 90 models, whose predicted class labels were combined and considered as reduced features. On the basis of these features, a multilayer perceptron was applied and developed the final prediction model (IML-TYLCVs). We conducted blind prediction on 3 groups using IML-TYLCVs, and the results indicated that 2 groups were severe and 1 group was mild. Furthermore, we confirmed the prediction with virus-challenging experiments of tomato plant phenotypes using infectious clones from 3 groups. Plant virologists and plant breeding professionals can access the user-friendly online IML-TYLCVs web server at https://balalab-skku.org/IML-TYLCVs, which can guide them in developing new protection strategies for newly emerging viruses.
@article{bupi2023effective, title = {An effective integrated machine learning framework for identifying severity of tomato yellow leaf curl virus and their experimental validation}, author = {Bupi, Nattanong and Sangaraju, Vinoth Kumar and Phan, Le Thi and Lal, Aamir and Vo, Thuy Thi Bich and Ho, Phuong Thi and Qureshi, Muhammad Amir and Tabassum, Marjia and Lee, Sukchan and Manavalan, Balachandran}, journal = {Research}, volume = {6}, pages = {0016}, year = {2023}, publisher = {AAAS}, doi = {10.34133/research.0016}, url = {https://spj.science.org/doi/10.34133/research.0016}, published = {true}, dimensions = {true}, }